
CompareTE
A pipeline to compare TE content across genomes using various platforms.
Requirements
- Nextflow >=26.04 (install)
- Java 11+ (required by Nextflow)
- Container engine — one of:
- Docker
- Singularity / Apptainer
- (or install all tools manually if not using containers)
The easiest approach is Docker + Nextflow on Linux/Mac, or Docker Desktop on Windows.
Quick Start
To run the pipeline immediately with test data:
nextflow run main.nf -profile docker,test_bacteria
This runs Earl Grey, HiTE, and RepeatMasker on three bacterial genomes using Docker. Results appear in results/.
For your own data, create a CSV file (see Input below) and run:
nextflow run main.nf --input your_input.csv -profile docker
Input
Input should be a samplesheet CSV file (ending in .csv) with no header row.
Each row contains: name,source where source is one of:
Option 1: RefSeq ID (downloaded automatically)
Escherichia_coli,GCF_000005845.2
Bacillus_subtilis,GCF_000005825.4
Option 2: Local genome file
Drosophila_melanogaster,/full/path/to/dm6.fa
Caenorhabditis_elegans,/full/path/to/ce11.fa
Option 3: Genome + annotation (annotation is optional)
Arabidopsis_thaliana,/full/path/to/TAIR10.fa,/full/path/to/TAIR10.gff3
File format requirements:
Genome must end with: .fa, .fasta, .fna, .fa.gz, .fasta.gz, or .fna.gz
Annotation (if provided) must end with: .gff, .gff3, .gff.gz, or .gff3.gz
⚠️ Do not use relative paths — use absolute paths only
See examples in data/ folder.
Running the pipeline
To run the pipeline you need to make a csv input file as described above with either Refseq IDs (these always begin GCF_…), or with genomes, or genomes and annotation files.
To run with all the different TE programs on a input csv file called input.csv:
nextflow run main.nf --orthofinder --hite --earlgrey --input input.csv
Though, the above would require you to manually download all the prerequisites and programs, so the easier way is to set a container engine to pull all the programs you need:
nextflow run main.nf --orthofinder --hite --earlgrey --input input.csv -profile docker/singularity/apptainer
Orthofinder (optional)
OrthoFinder v2.5.5 performs ortholog detection and generates a species tree. Use this if you want to build a phylogenetic tree or identify orthologous genes across your input genomes.
Requires genome annotation (GFF file in the samplesheet). Run with:
nextflow run main.nf --orthofinder --input your_input.csv -profile docker
Earl Grey runs from the tobybaril/earlgrey_dfam3.7 container, which already ships a fully-configured RepeatMasker with Dfam 3.7. So in most cases you just run:
nextflow run main.nf --earlgrey --input input.csv -profile singularity
If you want to use a newer Dfam release, pass --famdb pointing at a directory containing the famdb partitions (e.g. dfam39_full.N.h5 files):
nextflow run main.nf --earlgrey --famdb /path/to/dfam39 --input input.csv -profile singularity
--famdb is bind-mounted into the container at /opt/conda/share/RepeatMasker/Libraries/famdb, overriding the bundled Dfam. Note this replaces only the famdb, not the container’s prebuilt RepeatMasker library, so a mismatched Dfam version may cause issues — the bundled Dfam 3.7 is the safest default.
RepeatMasker / RepeatModeler (--repeatmasker)
An optional subworkflow annotates TEs with RepeatMasker, using a repeat library built from your --famdb Dfam database (via famdb.py). Optionally, --run_repeatmodeler first builds a de novo library per genome (slow) and merges it with the Dfam library. The library is de-duplicated with a clustering tool before masking.
nextflow run main.nf --repeatmasker --famdb /path/to/dfam39 --input input.csv -profile singularity
Options:
--run_repeatmodeler— also build a de novo library with RepeatModeler (adds substantial runtime per genome).--te_clusterer—linclust(default),mmseqs, orcdhit.--repeatmasker_speed—qq(fastest, default),q, ordefault(most sensitive).--famdb_lineage— lineage to extract from the famdb (e.g.hymenoptera); defaults toroot.
Outputs (masked genome, .out, .tbl, .gff, the repeat library, and a combined .tsv) are written to results/repeatmasker/.
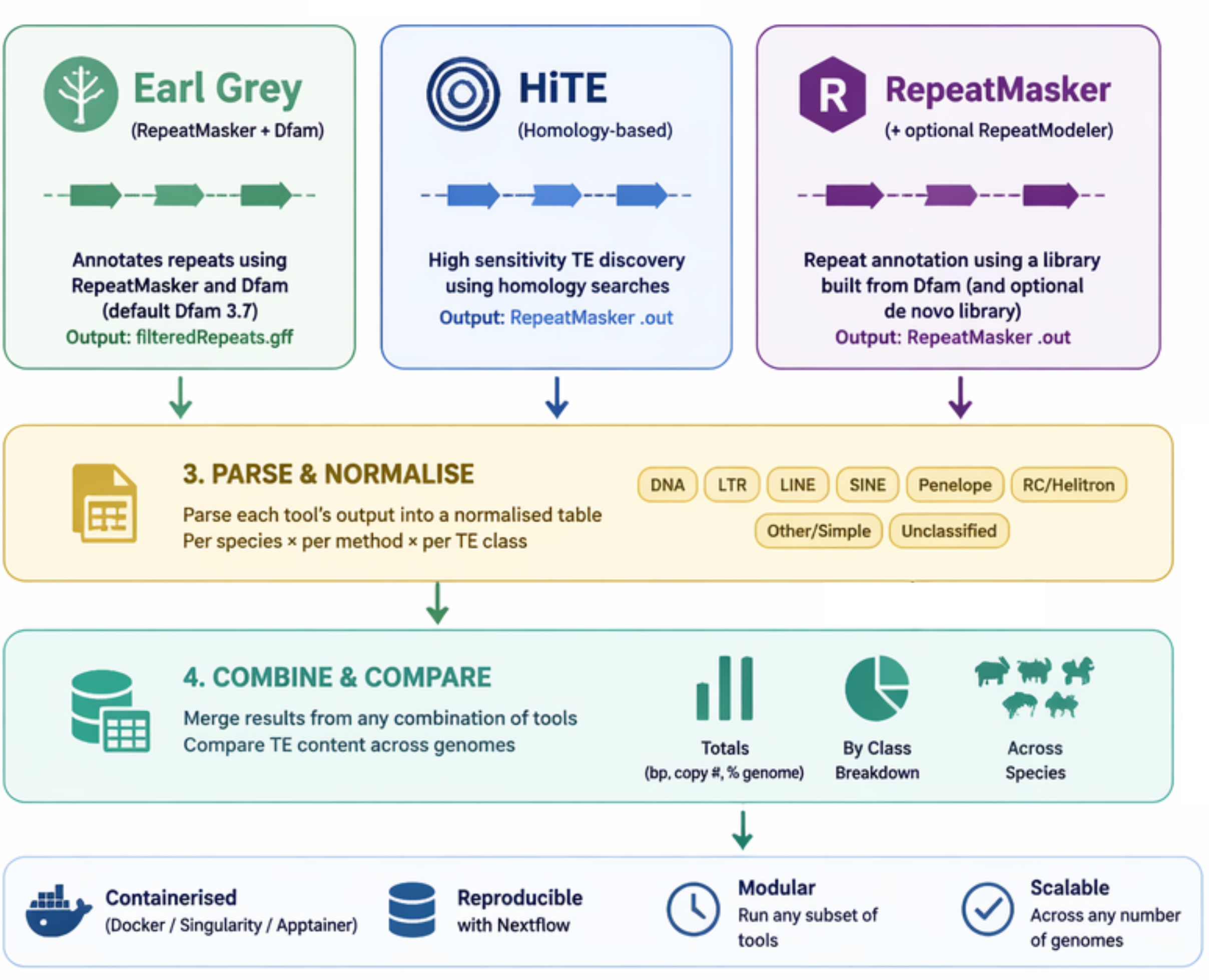
Combining TE methods (PARSE_TE / COMBINE_TE)
Whenever one or more TE methods are run (--earlgrey, --hite, --repeatmasker), the pipeline parses each method’s output into a normalised per-species, per-method table (PARSE_TE) and then merges everything into combined outputs (COMBINE_TE). It reads Earl Grey’s filteredRepeats.gff, HiTE’s RepeatMasker .out, and RepeatMasker’s .out, mapping all of them to common high-level TE classes (DNA, LTR, LINE, SINE, Penelope, RC/Helitron, Other/Simple, Unclassified).
This works for any subset of methods — one, two, or all three. Outputs in results/compare_te/:
combined_te_totals.tsv— total TE bp, copy number, and % genome for every species × method.combined_te_by_class.tsv— per-class breakdown for every species × method.combined_te_comparison.pdf— a cross-species totals bar chart (coloured by method) and a per-class figure faceted by species.
nextflow run main.nf --earlgrey --hite --repeatmasker --famdb ... --input input.csv -profile singularity
HPC cluster profiles
Bundled institutional profiles configure the scheduler, container engine, and resource limits for a specific cluster. They already enable the right container engine, so you do not need to add -profile singularity or an external -c config.
| Profile | Cluster | Scheduler | Container |
|---|---|---|---|
ucl_myriad |
UCL Myriad | SGE | Singularity/Apptainer |
cambridge |
University of Cambridge CSD3 | SLURM | Singularity |
Combine an HPC profile with a test or input profile, e.g. on UCL Myriad:
nextflow run main.nf -profile ucl_myriad,test_bacteria -resume --hite
On Cambridge CSD3, export your SLURM project/account first (defaults to the icelake partition):
export NXF_CAMBRIDGE_PROJECT=MYPROJECT-SL2-CPU
nextflow run main.nf -profile cambridge,test_bacteria -resume --hite
Useful additional flags:
-resume : This allows the pipeline to resume from the last failed process (using the nextflow cache-ing mechanism)
-bg : This allows nextflow to run in the background, so you can continue to use your terminal.
Current test commands:
nextflow run main.nf -profile docker,test_bacteria -resume
To run with HITE:
nextflow run main.nf -profile docker,test_bacteria -resume --hite
To run with EARL GREY:
nextflow run main.nf -profile docker,test_bacteria -resume --earlgrey
To run orthofinder on your input species:
nextflow run main.nf -profile docker,test_bacteria -resume --orthofinder
Test a docker container:
docker run -it --volume $PWD:$PWD <container> bash